Blog cerrado
A partir de este momento, el blog de Docking Proteínas quedará cerrado.
Podrán seguir las noticias a través de la web oficial de Ibercivis.
Disculpen las molestias.
A partir de este momento, el blog de Docking Proteínas quedará cerrado.
Podrán seguir las noticias a través de la web oficial de Ibercivis.
Disculpen las molestias.
Hola de nuevo docker@s!
Diversos artículos aparecidos en prensa sobre Ibercivis, en concreto el proyecto de docking, hacen mención a que esta plataforma ayudará en la lucha contra el cáncer gracias a las moléculas que se encuentren. Por desgracia, dicha enfermedad convive con nosotros día a día, ya sea directamente o a través de alguien cercano, por lo que todos tenemos una idea aproximada de lo que es. Pero, ¿qué es realmente el cáncer?
El cáncer consiste en el crecimiento descontrolado y diseminación de células anormales en el organismo. Dichas células invaden y dañan tejidos y órganos. Todos los cánceres se originan como consecuencia de mutaciones en los genes de nuestras células, siendo por tanto una enfermedad genética. El cáncer es la segunda causa de muerte en los países desarrollados, en los que una de cada cuatro personas fallece debido a esta enfermedad. En España, 82.000 personas mueren cada año como consecuencia del cáncer. La carcinogénesis o aparición de un cáncer es el resultado de dos procesos sucesivos: el aumento descontrolado de la proliferación de un grupo de células que da lugar a un tumor o neoplasia, y la posterior adquisición por estas células de capacidad invasiva, que les permite diseminarse desde su sitio natural en el organismo y colonizar y proliferar en otros tejidos u órganos (proceso conocido como metástasis).
El tratamiento del cáncer se basa en tres pilares fundamentales, que son la cirugía, la quimioterapia y la radioterapia (combinada o no con hormonoterapia), que pueden administrarse solos o en asociación. Además, se utilizan otros tratamientos, como el transplante de médula ósea, la inmunoterapia o la terapia génica.
La quimioterapia consiste en el uso de medicamentos antineoplásicos para tratar las células cancerosas. Se ha utilizado durante muchos años y es uno de los tratamientos más comunes contra el cáncer. En la mayoría de los casos, la quimioterapia actúa interfiriendo con la capacidad de crecimiento o reproducción de las células cancerosas. Distintos grupos de medicamentos actúan en forma diferente para combatir las células cancerosas. A menudo, se utiliza una combinación de medicamentos quimioterapéuticos para combatir una clase específica de cáncer.
Los medicamentos quimioterapéuticos, antineoplásicos o citotóxicos, actúan sobre las células tumorales impidiendo su proliferación. La mayor parte de medicamentos actúan sobre el ciclo celular, interfiriendo en la síntesis del ADN y del ARN o inhibiendo la maquinaria celular que hace posible que se sinteticen nuevos elementos para formar de nuevo células tumorales. Actualmente, en el tratamiento quimioterapéutico pueden utilizarse más de 50 medicamentos para combatir el cáncer y prevenir el crecimiento, la multiplicación y la diseminación de las células cancerosas. Si bien la quimioterapia puede ser bastante eficaz en el tratamiento de ciertos cánceres, los medicamentos quimioterapéuticos alcanzan todas las partes del cuerpo, no sólo las células cancerosas. Por este motivo, es posible que surjan diversos efectos secundarios durante el tratamiento.
Otro problema que puede surgir es el de la posible resistencia a la quimioterapia mediante una respuesta celular a los ataques citotóxicos al ADN. Estas respuestas incluyen la inducción de la muerte celular, la modulación de la progresión del ciclo celular, la tolerancia al daño y la iniciación de la reparación del ADN. Estos mecanismos de defensa producen un efecto negativo en la eficacia de los tratamientos quimioterapéuticos. Es por ello que la inhibición farmacológica de estas respuestas potenciaría la acción citotóxica de un determinado rango de agentes anticancerígenos. Así pues la búsqueda de estos inhibidores es hoy en día una de las metas para mejorar los tratamientos de quimioterapia.
Uno de los activos más importantes en la célula para la reparación de daños en el ADN es la proteína humana O6-alkylguanine DNA alkyltransferase (MGMT o hAGT), ya que se encarga de proteger al ADN de los agentes dañinos del entorno, por lo que juega un importante papel como mecanismo de resistencia a determinados tratamientos contra el cáncer. Lo sabemos debido a que se ha detectado que las células tumorales frecuentemente expresan una gran cantidad de MGMT, y se ha observado en diferentes tipos de cáncer: de colon, tumores de pulmón, de mama, tumores pancreáticos, linfomas, mielomas y gliomas entre otros. Debido a esto, la inhibición farmacológica de MGMT potenciaría el efecto citotóxico de un diverso abanico de agentes anticancerígenos, especialmente en tumores de colon y cerebrales.
Pero a pesar del interés farmacológico que ha despertado esta proteína, solo unas cuantas moléculas se han desarrollado como inhibidores, ninguna de las cuales tiene el grado de efectividad deseado. Es por ello que este proyecto ha comenzado con la búsqueda de inhibidores de MGMT.
Si queréis más información sobre el cáncer, aquí tenéis el enlace al Centro Nacional de Investigaciones Oncológicas (CNIO), donde tienen una página muy buena sobre el tema:
http://cancernet.nci.nih.gov/cancertopics/understandingcancer/espanol/cancer/
Un saludo a todos!
Equipo de Docking
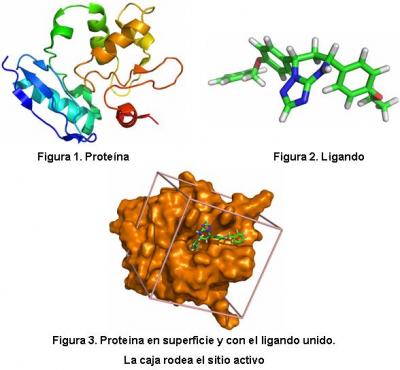
Hola docker@s!
Bueno, supongo que alguno de vosotros os estaréis preguntando cómo funciona más o menos esto del docking. Pues bien, para empezar llamamos así al proceso de unión de dos moléculas (proteínas, ADN, ARN, moléculas pequeñas...). Conocer qué moléculas se unen y de qué manera resulta muy importante para entender las diversas funciones biológicas del organismo. En nuestro caso nos centramos en las interacciones entre proteínas (Figura 1) y moléculas pequeñas (a las que llamamos genéricamente ligandos, Figura 2) ya que la mayoría de los fármacos están basados en este tipo de uniones. Conociendo la estructura de una determinada proteína es posible encontrar ligandos que puedan modificar dicha función, ya sea inhibiéndola o potenciándola según la necesidad que se tenga.
Teniendo identificada una proteína cuya función se quiere modificar, ahora viene el tema complicado: ¿cuál es el ligando que hace lo que queremos? Saber si un ligando puede unirse a una proteína no es una tarea trivial, y si a esto le sumamos el hecho de que el número de posibles ligandos tiende a infinito, el tema se complica aún más. Así pues lo que hacemos es partir de un conjunto de ligandos (quimioteca) que están disponibles comercialmente a través de diferentes proveedores. Aún así, siguen siendo muchos, en estos momentos tenemos más de 4 millones.
Probar todos estos ligandos con nuestra proteína en un laboratorio experimental llevaría demasiado esfuerzo (podría incluso ser inabordable) tanto a nivel económico como de mano de obra y tiempo. Por lo tanto hacer una simulación por ordenador de la unión entre la proteína y los ligandos es una alternativa muy buena que puede ahorrar mucho tiempo y dinero.
Pero, ¿cómo se hace esta simulación? En primer lugar tenemos que convertir la información necesaria de la proteína y los ligandos a un tipo manejable por el ordenador. La preparación es un poco diferente para la proteína y para los ligandos:
Pues bien, con la proteína y los ligandos preparados, ahora toca el tema que computacionalmente resulta más costoso: simular el docking de cada ligando con la proteína y calcular como de buena es la interacción entre ambos. A este proceso se le llama cribado virtual de la quimioteca. Para ello se toma un ligando y se va trasladando y rotando por todo el centro activo, generando lo que llamamos poses. Cada una de estas poses se evalúa la energía de unión del complejo proteína-ligando que se forma. Cuanto menor es esta energía, tanto mejor es la interacción, y por tanto más estable será el complejo formado. Un solo docking (la proteína y 1 ligando) puede tardar alrededor de 5 minutos. Con tiempos así se emplearían alrededor de 40 años en procesar los 4 millones de moléculas. Pero gracias al proyecto Ibercivis y a vuestra participación es posible tenerlo en un tiempo muchísimo más razonable.
Cuando el cribado virtual está completo, tomamos los resultados de cada docking y los insertamos en una base de datos para analizarlos. Se hace una ordenación según su grado de interacción con la proteína (energía). Adicionalmente, para la parte superior de la lista (por ejemplo los 100 primeros) realizamos procesos más precisos y a la vez más costosos computacionalmente (de ahí que se restrinja su número hasta 100), como por ejemplo Dinámica Molecular, con el fin de realizar un estudio más minucioso de las interacción. Los resultados de Dinámica Molecular por lo general producen una ordenación más fiable que la anterior. Finalmente, es siempre necesario realizar una inspección visual en el ordenador los mejores ligandos para su selección y adquisición.
Nuestros colegas experimentales son los encargados de llevar a cabo las pruebas en su laboratorio mediante ensayos in vitro (en el tubo de ensayo) e in vivo (directamente en las células). Aquellos ligandos seleccionados que también funcionen experimentalmente se convierten en cabezas de serie que podrían en un futuro llegar a ser el principio activo de un nuevo fármaco.
Este es más o menos el proceso, iremos dando más detalles según vayáis solicitando más información o según vayamos haciendo cosas nuevas.
Un saludo a todos!
Equipo de Docking
Nuestro objetivo fundamental es encontrar nuevas moléculas que puedan convertirse en fármacos eficaces en la lucha contra el cáncer. Para ello trabajamos en el desarrollo de algoritmos cuya finalidad consiste en predecir cómo estas moléculas se unen a sus receptores biológicos y así poder, de alguna manera, modular el comportamiento de éste, por ejemplo, inhibiendo el ciclo reproductivo del ADN dañado, frenando por tanto la proliferación de las células cancerígenas.
La búsqueda de estas moléculas se realiza en grandes bases de datos que llegan a contener millones de ellas. Además, cada una de estas moléculas tiene una complejidad estructural inherente que se traduce en multitud de disposiciones tridimensionales distintas que es necesario tener en cuenta y probar. Esto hace que el número total de estructuras a evaluar sea virtualmente infinito.
Para poder realizar este tipo de experimentos es necesario disponer de potentes ordenadores, y cuantos más mejor. Es aquí donde proyectos como Ibercivis adquieren una relevancia especial.
Ibercivis nos va a permitir cribar virtualmente, que es como se conoce a la técnica que estamos desarrollando, millones de moléculas en tiempo record. Esto supone una aceleración muy importante en el proceso de diseño de nuevas moléculas, muy largo y costoso en la actualidad.
Los proyectos de computación voluntaria cumplen una doble misión. Por una parte representan una ayuda inestimable en cuanto a la cantidad de cálculos que podemos realizar. Por otra, los voluntarios entran en contacto directo con la investigación científica que se está realizando en su propio ordenador, adquiriendo nuevos conocimientos y mayor concienciación del problema a resolver.
Algunos proyectos similares que ya han obtenido resultados prometedores son el salva pantallas de la Universidad de Oxford dedicado al cáncer y WISDOM a la malaria. En el primer caso, y después de un año, se llegaron a usar alrededor de 1.5 millones de ordenadores en más de 200 países. Con Ibercivis esperamos obtener resultados competitivos.
Aunque a una escala sensiblemente menor en la Unidad de Bioinformática del CBMSO ya hemos empezado a obtener resultados importantes en un par de proyectos relacionados con el cáncer. En concreto hemos patentado varias moléculas con resultados experimentales positivos.
Gracias a todos por vuestra ayuda,
El equipo de Docking

Todos los medicamentos incluyen en su composición una sustancia química llamada principio activo (a la que genéricamente denominaremos ligando) y que es responsable de la actividad de dicho medicamento.
El resto de sus componentes, denominado excipiente, está constituido por sustancias inactivas cuya misión, entre otras, es asegurar que el principio activo llegue al lugar donde debe actuar.
Estos lugares normalmente se encuentran localizados tanto en la superficie como en el interior de ciertas estructuras macromoleculares "receptores" como son las proteínas y los ácidos nucleicos entre otros.
Al final de su viaje, un ligando ha de encontrar su centro activo y acoplarse a él. Este proceso, denominado docking, es bastante complejo y en él entran en juego una serie de procesos químicos gobernados por leyes de físicas, entre ellas las que tienen que ver con la energía que se consume o libera en tal proceso.
El saber cómo ocurre esta unión, así como la caracterización y cuantificación de los distintos eventos que tienen lugar en tal proceso, es un área de investigación en creciente desarrollo, ya que este conocimiento nos aportará los elementos necesarios que nos permitirá, en teoría, diseñar moléculas con la estructura óptima necesaria para que su actividad sea no sólo mucho mejor, sino que además no produzca interferencias con otros centros activos no deseados, lo que darían lugar a los conocidos "efectos secundarios".
En la actualidad contamos con sofisticadas técnicas experimentales de cuyos resultados se extrae información tridimensional tanto de las proteínas como de los ligandos, es decir, cómo están situados sus átomos en el espacio y cuál es la disposición geométrica de la unión entre ambos. Por lo tanto, sabemos cómo se han unido las moléculas y cuánto vale esa unión. Si fuéramos capaces, a partir de esas estructuras tridimensionales y con las leyes físicas y químicas que conocemos, de reproducir los resultados experimentales, estaríamos en la posición de poder predecir, para cualquier otro ligando, cómo sería su unión y cuánto valdría, antes de hacer costosas pruebas farmacológicas.