¿Cómo funciona el docking?
Hola docker@s!
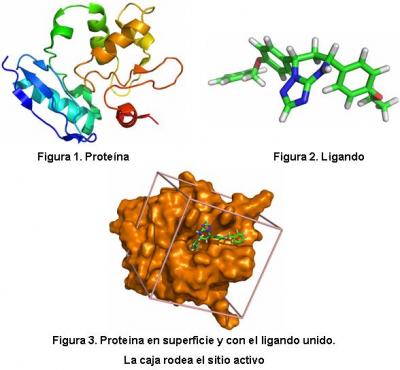
Bueno, supongo que alguno de vosotros os estaréis preguntando cómo funciona más o menos esto del docking. Pues bien, para empezar llamamos así al proceso de unión de dos moléculas (proteínas, ADN, ARN, moléculas pequeñas...). Conocer qué moléculas se unen y de qué manera resulta muy importante para entender las diversas funciones biológicas del organismo. En nuestro caso nos centramos en las interacciones entre proteínas (Figura 1) y moléculas pequeñas (a las que llamamos genéricamente ligandos, Figura 2) ya que la mayoría de los fármacos están basados en este tipo de uniones. Conociendo la estructura de una determinada proteína es posible encontrar ligandos que puedan modificar dicha función, ya sea inhibiéndola o potenciándola según la necesidad que se tenga.
Teniendo identificada una proteína cuya función se quiere modificar, ahora viene el tema complicado: ¿cuál es el ligando que hace lo que queremos? Saber si un ligando puede unirse a una proteína no es una tarea trivial, y si a esto le sumamos el hecho de que el número de posibles ligandos tiende a infinito, el tema se complica aún más. Así pues lo que hacemos es partir de un conjunto de ligandos (quimioteca) que están disponibles comercialmente a través de diferentes proveedores. Aún así, siguen siendo muchos, en estos momentos tenemos más de 4 millones.
Probar todos estos ligandos con nuestra proteína en un laboratorio experimental llevaría demasiado esfuerzo (podría incluso ser inabordable) tanto a nivel económico como de mano de obra y tiempo. Por lo tanto hacer una simulación por ordenador de la unión entre la proteína y los ligandos es una alternativa muy buena que puede ahorrar mucho tiempo y dinero.
Pero, ¿cómo se hace esta simulación? En primer lugar tenemos que convertir la información necesaria de la proteína y los ligandos a un tipo manejable por el ordenador. La preparación es un poco diferente para la proteína y para los ligandos:
- Proteína: tenemos que delimitar lo que se denomina centro activo, es decir, la zona de la proteína donde ésta interacciona con los ligandos (Figura 3). Esta información, en la mayor parte de los casos en los que trabajamos, está disponible.
- Ligandos: por lo general, una molécula viene representada por lo que se llaman conformaciones, es decir, distinta disposición espacial de los elementos que la forman. Esto es debido a que los ligandos no son rígidos, sino que hay ciertos enlaces (uniones entre los átomos) que tienen la capacidad de girar. Por supuesto a las proteínas les ocurre lo mismo, pero por ahora estas se considerarán como rígidas. La inclusión de la flexibilidad de la proteína dentro de los algoritmos de docking no es nada trivial, y es de hecho un área de investigación muy activa.
Pues bien, con la proteína y los ligandos preparados, ahora toca el tema que computacionalmente resulta más costoso: simular el docking de cada ligando con la proteína y calcular como de buena es la interacción entre ambos. A este proceso se le llama cribado virtual de la quimioteca. Para ello se toma un ligando y se va trasladando y rotando por todo el centro activo, generando lo que llamamos poses. Cada una de estas poses se evalúa la energía de unión del complejo proteína-ligando que se forma. Cuanto menor es esta energía, tanto mejor es la interacción, y por tanto más estable será el complejo formado. Un solo docking (la proteína y 1 ligando) puede tardar alrededor de 5 minutos. Con tiempos así se emplearían alrededor de 40 años en procesar los 4 millones de moléculas. Pero gracias al proyecto Ibercivis y a vuestra participación es posible tenerlo en un tiempo muchísimo más razonable.
Cuando el cribado virtual está completo, tomamos los resultados de cada docking y los insertamos en una base de datos para analizarlos. Se hace una ordenación según su grado de interacción con la proteína (energía). Adicionalmente, para la parte superior de la lista (por ejemplo los 100 primeros) realizamos procesos más precisos y a la vez más costosos computacionalmente (de ahí que se restrinja su número hasta 100), como por ejemplo Dinámica Molecular, con el fin de realizar un estudio más minucioso de las interacción. Los resultados de Dinámica Molecular por lo general producen una ordenación más fiable que la anterior. Finalmente, es siempre necesario realizar una inspección visual en el ordenador los mejores ligandos para su selección y adquisición.
Nuestros colegas experimentales son los encargados de llevar a cabo las pruebas en su laboratorio mediante ensayos in vitro (en el tubo de ensayo) e in vivo (directamente en las células). Aquellos ligandos seleccionados que también funcionen experimentalmente se convierten en cabezas de serie que podrían en un futuro llegar a ser el principio activo de un nuevo fármaco.
Este es más o menos el proceso, iremos dando más detalles según vayáis solicitando más información o según vayamos haciendo cosas nuevas.
Un saludo a todos!
Equipo de Docking
15 comentarios
juan mateus -
jordan 12 -
maicol -
Jorge -
Me pareció muy interesante la información que muestras. Necesito dar un seminario y he estado traduciendo un review sobre estudios docking pero es mucha la información que contiene. Me gustaría que me sugirieras algunos link en español donde pueda porfundizar la información. Saludos
malen burgos -
Horacio -
Antonio Morreale -
La verdad es que acabas de preguntar por un tema un tanto complejo, no tanto por la dificulatad teórica sino más bien por su implementación,
de una manera eficaz, en un programa de docking.
Es un tema que está generando muchísimos artículos, pero que desafortunadamente no tiene, hoy por hoy, una respuesta sencilla.
Nosotros en particular no tenemos en cuenta el efecto de la flexibilidad del receptor al hacer docking, aunque al final de todo
el protocolo de cribado virtual realizamos simulaciones de dinámica molecular con el fin de mejorar el ajuste entre el ligando y el receptor.
Las formas de incorporar la flexibilidad del receptor en docking son muy distintas y van desde aproximaciones muy sencillas
(atenuación del potencial de interacción de van der Waals) hasta muy complejas (modos normales esenciales).
También depende mucho del número de moléculas con las que quieras realizar el docking, si existe o no estructuras cristalizadas de compuestos
análogos que te puedan servir como guía para las tuyas...
En resumen, es un tema muy complejo y la respuesta no es trivial.
Como te decía, hay abundante bibliografía sobre el tema (algunas revisiones te las puedo mandar para que veas cómo está el tema). Además, si quieres,
a modo personal puedes contactar conmigo a través del correo electrónico (amorreale@cbm.uam.es) por si quieres algo más detallado.
Un saludo y gracias por usar Ibercivis.
Antonio Morreale
Equipo de Docking
Luis -
Antonio Morreale -
Con respecto a tu primera pregunta, aún es pronto para contestar a esta pregunta, ten en cuenta que no hace mucho que se ha iniciado el proyecto. De todas formas tenemos previsto en breve hacer un análisis de los resultados que ya tenemos. Os lo haremos saber tan pronto como los tengamos. Con respecto al "modelo gráfico" por el que preguntas, estamos en ello, y también en breve esperamos tenerlo a vuestra disposición.
Un saludo y gracias por unirte a Ibercivis.
Antonio Morreale
Equipo de docking
Marcos -
1) ¿hay alguna novedad importante en los resultados que os han estado llegando estos dias?
2) ¿habrá algún "modelo gráfico" mietras ejecutas el proyecto(como lo tiene seti@home o climatteprediction,x ejemplo.)?????
saludos
Ruben Gil Redondo -
En la vista simple te aparece materiales64 porque es la aplicacion que esta ejecutandose en ese momento. Como sabes Ibercivis consta de tres proyectos cientificos (materiales, fusion y docking) que se distribuyen en el tiempo de calculo.
El de docking es el Proteinas. Si solo quieres hacer tareas de este tipo (o del tipo de los otros proyectos) puedes configurarlo desde la pagina de Ibercivis: Menu Ibercivis y despues en Tu Cuenta. Ahi en el menu Subproyectos puedes dejar marcados aquellos que quieres que se ejecuten en tu ordenador.
Venga espero que te haya sido de ayuda. Un saludo.
Ruben
Rubentronics -
En la ventana de "vista simplificada" del Boinc me aparece el proyecto "materiales64" en lugar de "proteínas". ¿Qué debo cambiar?
Rompeolas -
Estaría bien que cuando se consiga probar toda una quimioteca sobre una proteína se comunicara y bueno, cuando hayan otros resultados no me cabe duda que nos avisaréis ^_^
Un saludo.
Atte. Rompeolas CANAL@Boinc
galle -
sería espectacular tener una visualización del proceso al procesar las wus 8) tal vez cuando hayan terminado de retocar el proyecto podrían intentarlo...
Apa -